Quickstart
Let's start with an easy example of what a typical use case would be like. We'll be calculating the volume of the main cavity of an Odorant Binding Protein along a very short trajectory. You can get all files used in this example here.
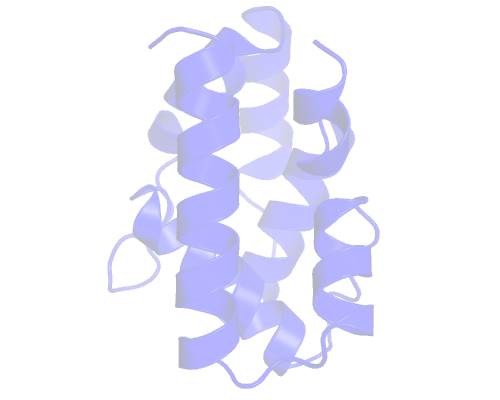
Discovering pockets
The pocket we are after is located in the center, delimited by those α-helices. The exact definition of a pocket is hard to define but we'll do our best.
First, let's run ANA naively, without any configuration file:
ANA2 input_pdb.pdb -f 1_cavity
Pocket 253.536
Pocket 38.5048
The input structure always go first, so you won't need to write its flag, this
is the only mandatory argument when running ANA. The flag -f is the shorthand
for the --output_draw argument. 1_cavity will be the prefix for all
the output cavities ANA finds.
In this case we get 2 output files, each with a different pocket. If we load them on pymol (or any other biomolecule visualizer), along with the input structure, we'll see something like this:
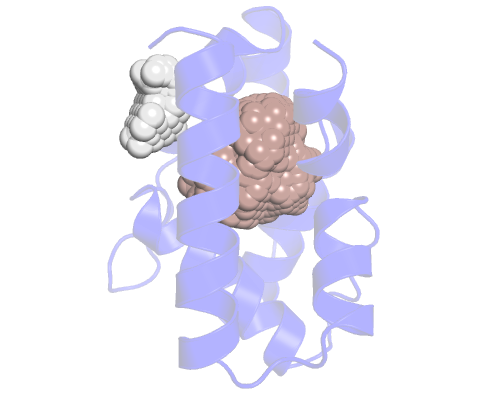
The first pocket (the salmon colored one) is close to the one we want, so we'll choose that one. We can also guess there should be more empy space under it but for some reason ANA is not showing it. We'll see how to fix this.
Defining our pocket
In order to draw an Included Area to track this pocket, we need the residues that surround it and a first approach to get them is to get what ANA calls wall residues or wall atoms. In order to get them, we have to write our first configuration file. ANA configuration files are just plain text files where each line is composed of an option, followed by a '=' character, and the appropriate arguments. So we will create a new file, write the following contents in it and call it config_1.cfg. The .cfg extension tells the VIM editor this is a config file, so VIM can provide appropriate highlighting. This is a nice-to-have, but is not necessary, ANA can't tell the difference.
list_wall = residue
separator = +
The residue keyword tells ANA we want a list of amino acids, the alternative would be to use the atom keyword, to get every atom that is in contact with the cavity. We don't need that level of detail, so we'll stay with the residues.
So, we will get a list of residues lining each cavity and each residue will be separated with the character '+'. We'll see later why we specified '+' as the separator character. Let's run it:
ANA2 input_pdb.pdb -c config_1.cfg -f 1_cavity -w wall_+
As you can see, we have 2 new arguments. -c or --config_file is the flag
for the configuration file and the -w or --output_wall tells ANA the name of
the text file that will contain the cavities lining residues.
Thus, we get the output file called wall_input_pdb. This is the section that matters to us:
pocket_1 Frame: 1
RESN | LEU+ILE+LEU+PHE+ILE+VAL+MET+LEU+VAL+VAL+SER+LYS+MET+VAL
RESI | 10+50+51+54+56+71+72+75+104+107+108+111+113+116
-------------
We can now copy the residue list and see them on pymol. Pymol needs the residue list to be separated with '+' characters, like this:
select resi 10+50+51+54+56+71+72+75+104+107+108+111+113+116
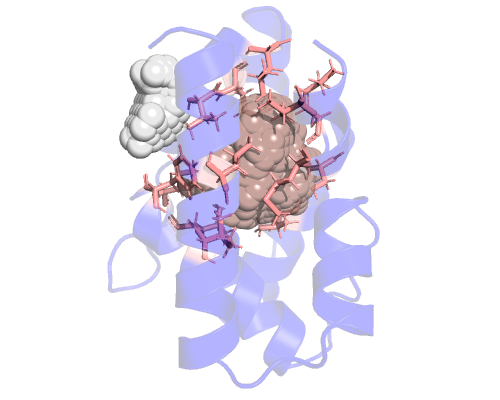
And that's why setting the separator variable is useful. If you want a more readable (tab separated) list you can simply remove that line in the config file or just comment it out. I created a new configuration file called config_2.cfg:
list_wall = residue
#separator = +
and re-run it:
ANA2 input_pdb.pdb -c config_2.cfg -f 1_cavity -w wall_
And now we have a new file, wall_ with a nicer output:
pocket_1 Frame: 1
RESN | LEU ILE LEU PHE ILE VAL MET LEU VAL VAL SER LYS MET VAL
RESI | 10 50 51 54 56 71 72 75 104 107 108 111 113 116
-------------
Now, let's paste this list in a new configuration file (config_3.cfg) to draw an Included Area.
included_area_residues = 10 50 51 54 56 71 72 75 104 107 108 111 113 116
and run it using the --tool_check_CH or -t flag. This will draw the convex
hull obtained by triangulating the alpha carbons from the residues listed in the
included_area_residues variable. Note that whenever we use this flag, ANA
will skip the cavity calculation and just calculate the Included Area Convex
Hull and write it into a PDB file with the name of our choosing, in this case:
ch_3_testing.pdb.
ANA2 input_pdb.pdb -c config_3.cfg -f 1_cavity -t ch_3_testing
If we load the ch_3_testing.pdb file on pymol, we'll see something like this:
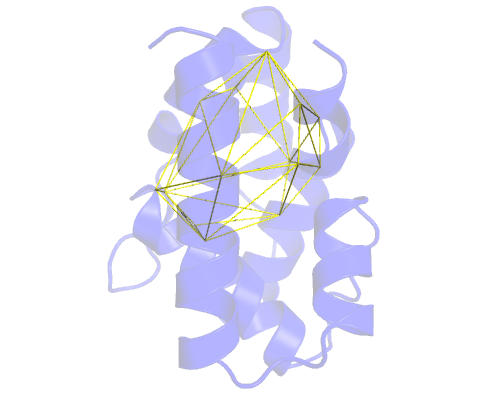
Refining our pocket
Now, as we said before, this was not exactly the pocket we were looking for. We have to modify the list of residues to define a proper cavity using the convex hull method.
It does not take long to get a sense of the resulting convex hull, given a set of points (alpha carbons, in our case). Whenever you see a problematic vertex, remove it from the list. And if you want to include an area that's being discarded, include a few of the residues that surround it.
Also, you don't want too many unnecessary residues listed in the included_area_residues variable since this will result in a minor, though unnecessary, overhead.
After pruning the list (removing several residues and adding new ones to get a more elongated cavity), we are left with this new config file, config_4.cfg:
included_area_residues = 10 17 27 43 54 57 71 72 75 81 89 99 107 108 111 113 116
included_area_precision = 1
As you can see, there's a new option: included_area_precision. When this
option is set to 0, ANA will triangulate the whole molecule and then discard
the tetrahedrons that don't intersect the Included Area. But if it's set to
1, it will also perform intersections between the Included Area and those
tetrahedrons that are not completely enclosed by the Included Area. For more
info about this, check the relevant paper.
We re-run the check to see the resulting convex hull:
ANA2 input_pdb.pdb -c config_4.cfg -f 1_cavity -t ch_4_testing
and load the ch_4_testing.pdb file on pymol:

Tracking our pocket along a MD run
We're now ready to track our pocket along a Molecular Dynamics trajectory. Our
trajectory is in the Amber NetCDF format, but ANA can read from other popular
formats such as .gro, .trr or .xtc.
We use the -d flag (think of dynamics) to include the trajectory and we
remove the -t flag, since this option will cancel every other calculation.
ANA2 input_pdb.pdb -c config_4.cfg -d input_trajectory.nc -o 5_cavity
Frame 1 87.2512
Frame 2 54.1299
Frame 3 56.8417
Frame 4 52.983
Frame 5 81.8424
Frame 6 69.003
Frame 7 43.4421
Frame 8 42.6606
Frame 9 50.9236
Frame 10 95.8178
Frame 11 79.5727
Frame 12 43.422
Frame 13 63.2107
Frame 14 55.336
Frame 15 62.7068
Frame 16 36.4506
Frame 17 64.9357
Frame 18 64.1257
Frame 19 44.2278
Frame 20 43.2665
Writing output...
Done
And now 5_cavity.pdb is a trajectory file with our pocket dynamics.
Using the -t flag you can see how the Included Area changed during the
trajectory, following the listed residues.
ANA2 input_pdb.pdb -c config_4.cfg -d input_trajectory.nc -f cavity -t ch_5_testing
The output of -t is also a trajectory file (in the PDB format). If we
load it, we can see the differences between the original (purple) static convex
hull and the dynamic one (green) from a random frame. In sand colored cartoon
representation, you can see our protein from the same random frame from the
trajectory:
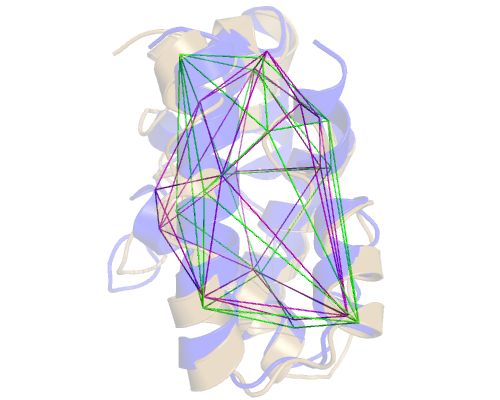
This trajectory came from a long Constant pH Replica Exchange MD, so the structure has experienced significant changes. Nevertheless, ANA's Included Area will follow the residues along the trajectory and update the Convex Hull at each frame.
Now you can play with the files if you want. You can change the cavity definition if you don't agree with mine and re-run the calculation. The good news is that, given a longer than 10 frames trajectory, (which is usually the case) there won't be much difference.
Addendum
Remember that whenever you use a tool like --tool_check_CH, ANA will only perform that task and exit, so you won't get any output other than the output from that particular tool.
You can redirect ANA's volume output to a file like this:
> ANA2 input_pdb.pdb -c config_4.cfg -d input_trajectory.nc -f 5_cavity -o output_vol
Writing output...
Done